

 2025-07
2025-07

1. 概述
对于药品质量标准的内容,从事分析工作的同学再熟悉不过,性状、鉴别、检查、含量测定。其中检查项基本可分为安全性检查和有效性检查,如下:
有效性检查:项目包括大家熟知的可见异物、不溶性微粒、水分、装量差异、溶出度、溶出曲线、含量均匀度、残留溶剂、有关物质等等......
安全性检查:微生物限度检查和无菌检查。
安全性检查是比有效性检查的项目更加需要关注和严格制定的。
那么,什么情况下质量标准中该制定微生物限度?什么情况下该制定无菌检查?限度又该如何确定?
2. 什么是微生物限度检查?
微生物限度(药典中常见的格式表述) 取本品,照非无菌产品微生物限度检查:微生物计数法(通则1105)和控制菌检查法(通则1106)及非无菌药品微生物限度标准(通则1107)检查,应符合规定。
微生物限度检查法:系检查非规定灭菌制剂及其原料、辅料受微生物污染程度的方法。检查项目包括需氧菌总数、霉菌和酵母菌总数及控制菌检查。
微生物计数法:用于能在有氧条件下生长的嗜温细菌和真菌的计数。其供试品检查包括三种方法:平皿法(包括倾注法和涂布法)、薄膜过滤法、MPN法,都是根据计数方法适用性试验确定的。MPN法用于微生物计数时精确度较差,但对于某些微生物污染量很小的供试品,MPN法可能是更适合的方法。
控制菌检查法:用于在规定的试验条件下,检查供试品中是否存在特定的微生物。根据药物剂型的不同,包括大肠埃希菌、耐胆盐革兰氏阴性菌、沙门菌、铜绿假单胞菌、金黄色葡萄球菌、梭菌、白色念珠菌。
计数法适用性试验:进行产品微生物限度检查时,应进行计数方法适用性试验,以确认采用的方法适合于该产品的微生物限度检查。进行供试液制备,然后接种和稀释,各试验菌应逐一进行微生物回收试验,微生物的回收可采用平皿法(包括倾注法和涂布法)、薄膜过滤法、MPN法。计数方法适用性试验中,釆用平皿法或薄膜过滤法时,试验组菌落数减去供试品对照组菌落数的值与菌液对照组菌落数的比值应在0.5~2范围内;采用MPN法时,试验组菌数应在菌液对照组菌数的95%置信限内。若各试验菌的回收试验均符合要求,照所用的供试液制备方法及计数方法进行该供试品的需氧菌总数、霉菌和酵母菌总数计数。
培养基适用性检查和控制菌检查方法适用性试验:供试品控制菌检查中所使用的培养基应进行适用性检查。供试品的控制菌检查方法应进行方法适用性试验,以确认所采用的方法适合于该产品的控制菌检查。
供试品检查:按计数方法适用性试验确认的计数方法进行供试品中需氧菌总数、霉菌和酵母菌总数的测定。胰酪大豆胨琼脂培养基或胰酪大豆胨液体培养基用于测定需氧菌总数;沙氏葡萄糖琼脂培养基用于测定霉菌和酵母菌总数。
结果判断
需氧菌总数:指胰酪大豆胨琼脂培养基上生长的总菌落数(包括真菌菌落数)。
霉菌和酵母菌总数:指沙氏葡萄糖琼脂培养基上生长的总菌落数(包括细菌菌落数)。若因沙氏葡萄糖琼脂培养基上生长的细菌使霉菌和酵母菌的计数结果不符合微生物限度要求,可使用含抗生素(如氯霉素、庆大霉素)的沙氏葡萄糖琼脂培养基或其他选择性培养基(如玫瑰红钠琼脂培养基)进行霉菌和酵母菌总数测定。
控制菌检查:阳性对照试验应检出相应控制菌、阴性对照试验应无菌生长,检查结果以供试品检查结果为准。
除中药饮片外,非无菌药品的需氧菌总数、霉菌和酵母菌总数照“非无菌产品微生物限度检查:微生物计数法(通则1105)”检查;非无菌药品的控制菌照“非无菌产品微生物限度检查:控制菌检查法(通则1106)”检查。各品种项下规定的需氧菌总数、霉菌和酵母菌总数标准解释如下∶
101cfu:可接受的最大菌数为20;
102cfu:可接受的最大菌数为200;
103cfu:可接受的最大菌数为2000;依此类推。
中药饮片的需氧菌总数、霉菌和酵母菌总数及控制菌检查照“中药饮片微生物限度检查法”(通则1108)检查;各品种项下规定的需氧菌总数、霉菌和酵母菌总数标准解释如下∶
101cfu:可接受的最大菌数为50;
102cfu:可接受的最大菌数为500;
103cfu:可接受的最大菌数为5000;
104cfu:可接受的最大菌数为50000;依此类推。
限度制定
微生物限度的制定,根据制剂、原料、中药提取物的不同,标准会有不同:
①制剂是非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准见表1:
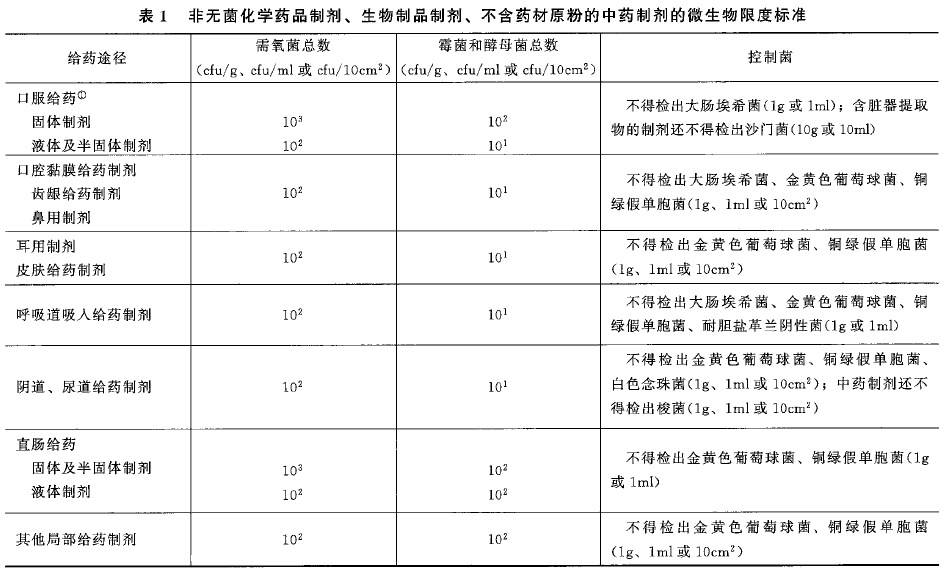
注:1.化学药品制剂和生物制品制剂若含有未经提取的动植物来源的成分及矿物质,还不得检出沙门菌(10g或10ml)。
2.特殊品种如透皮贴剂等,可采用贴为单位,限度标准以贴计。
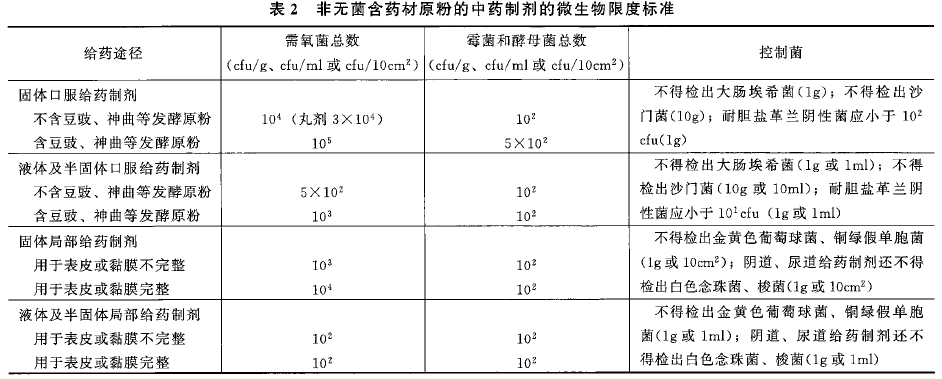
②非无菌含药材原粉的中药制剂的微生物限度标准见表2:
③非无菌药用原料及辅料的微生物限度标准见表3:

④中药提取物及中药饮片的微生物限度标准见表4:

3. 什么是无菌检查?
制剂通则或品种项下要求无菌及标示无菌的制剂和原辅料应制定无菌限度。包括注射剂、眼用制剂、吸入液体制剂、冲洗剂等,以及用于手术、严重烧伤、严重创伤的局部给药制剂。
无菌(药典中常见的格式) 取本品,经薄膜过滤法处理,依法检查(通则1101),应符合规定。
无菌检查法系用于检查药典要求无菌的药品、医疗器具、原料、辅料及其他品种是否无菌的一种方法。若供试品符合无菌检查法的规定,仅表明了供试品在该检验条件下未发现微生物污染。
无菌检查应在无菌条件下进行,试验环境必须达到无菌检查的要求,检验全过程应严格遵守无菌操作,防止微生物污染,防止污染的措施不得影响供试品中微生物的检出。单向流空气区域、工作台面及受控环境应定期按医药工业洁净室(区)悬浮粒子、浮游菌和沉降菌的测试方法的现行国家标准进行洁净度确认。隔离系统应定期按相关的要求进行验证,其内部环境的洁净度须符合无菌检查的要求。日常检验需对试验环境进行监测。
方法适用性试验 进行产品无菌检查时,应进行方法适用性试验,以确认采用的方法适合于该产品的无菌检查。进行菌种和菌液制备,采用薄膜过滤法或直接接种法进行适用性试验考察。
供试品的无菌检查 无菌检查法包括薄膜过滤法和直接接种法。只要供试品性质允许,应采用薄膜过滤法,包括水溶性液体供试品、醇类和油性供试品,或可在水或油性溶剂中溶解的供试品等。供试品无菌检查的所采用的检查方法和检验条件应与方法适用性试验确认的方法相同。
薄膜过滤法一般应采用封闭式薄膜过滤器。无菌检查用的滤膜孔径应不大于0.45μm,直径约为50mm。根据供试品及其溶剂的特性选择滤膜材质。使用时,应保证滤膜在过滤前后的完整性。
直接接种法适用于无法用薄膜过滤法进行无菌检查的供试品,即取规定量供试品分别等量接种至硫乙醇酸盐流体培养基和胰酪大豆胨液体培养基中。无菌检查时两种培养基接种的瓶或支数相等。除另有规定外,每个容器中培养基的用量应符合接种的供试品体积不得大于培养基体积的10%。
结果判断
供试品管 |
阴性管 |
阳性管 |
结果表明 |
+ |
- |
+ |
不符合规定 |
- |
- |
+ |
符合规定 |
+或- |
+ |
+ |
试验无效 |
+ |
- |
试验无效 |
|
- |
- |
试验无效 |
注:+表示有菌生长,-表示无菌生长 2025版《中国药典》1101 无菌检查法
4. 总结回顾
随便选一个药品原料、辅料、制剂,该做无菌?该做微生物限度?做哪方面内容?各自限度是什么?都能回答出来了吧?
大家可以通过下图回顾本文内容:
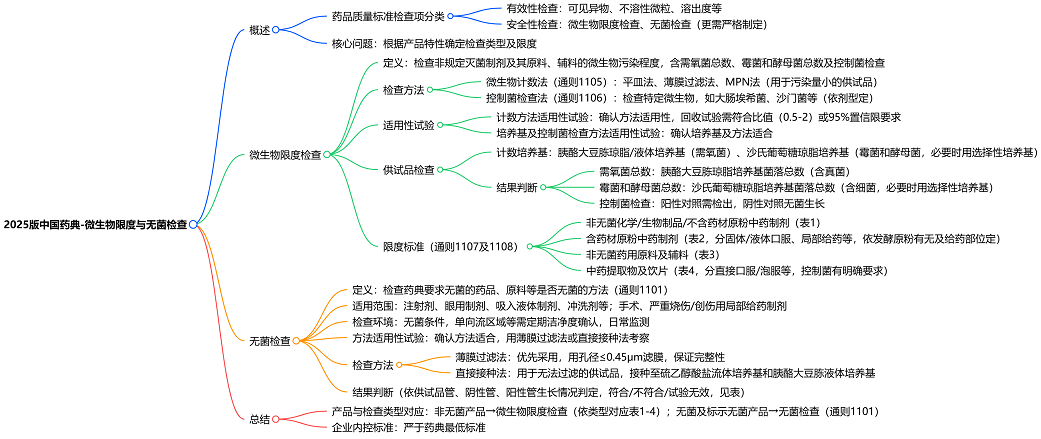
此外,药典标准毕竟是最低标准,企业会根据自己产品的具体质量制定内控标准,会比药典更严格一些。




