

 2026-05
2026-05

一、参比制剂遴选政策演进历程
发布时间 |
法规名称 |
主要内容 |
2015-08-18 |
(国务院关于改革药品医疗器械审怦审批制度的意见》(国发[2015]44号) |
提出“参比制剂由国家食品监管总局征询专家意见后确定,可选择原研药品,也可选择国际公认的同种药品” |
2016-03-08 |
普通口服固体制剂参比制剂选择和确定指导原则 |
提出了参比制剂选择原则。 |
2019-03-25 |
关于发布化学仿制药参比制剂遴选与确定程序的公告(2019年第25号) |
进一步明确了化学仿制药参比制剂遴选原则、遴选路径以及确定程序。 |
2020-10-19 |
国家药监局药审中心关于发布《化学仿制药参比制剂遴选申请资料要求》的通告(2020年第32号) |
制定标准化申报资料模板,明确证据链要求和真实性保障措施 |
2023-06-08 |
国家药监局关于发布化学仿制药参比制剂调整程序的公告(2023年第35号) |
建立动态调整机制,实现参比制剂目录的"有进有出",保障目录时效性和科学性 |
二、参比制剂遴选与确定程序
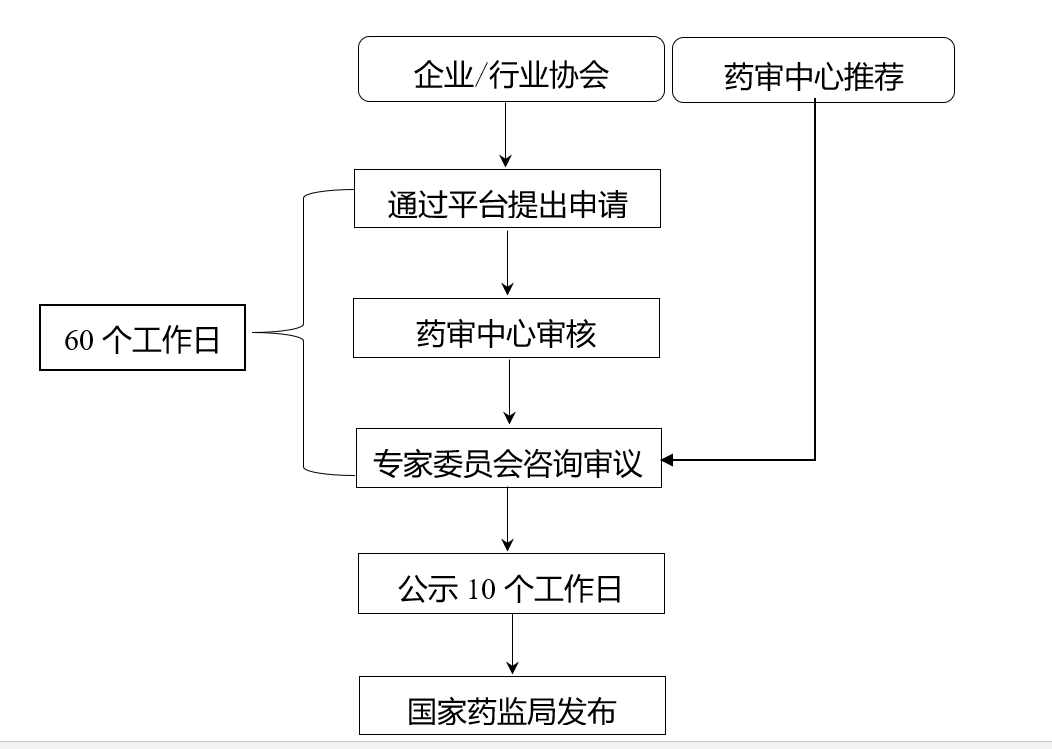
药品生产及研发企业或行业协会应按照上述原则,通过参比制剂遴选申请平台向国家药监局药品审评中心(以下简称药审中心)提出申请,药审中心在60个工作日内予以答复。
申请启动:药品生产企业、研发机构或行业协会通过CDE"参比制剂遴选申请平台"提交电子资料
形式审查:CDE在5个工作日内完成资料完整性审核,不符合要求的一次性告知补正
技术审评:药审中心组织药学、临床医学、统计学等领域专家进行技术审评,60个工作日内形成初步意见
结果公示:审议结果在CDE官网公示10个工作日,接受社会监督和异议反馈
异议处理:对公示期内的异议,CDE组织专家重新论证,必要时补充调研
目录发布:无异议或异议不成立的品种,由国家药监局正式纳入参比制剂目录
三、化学仿制药参比制剂遴选申请资料要求
根据2020-10-19国家药监局药审中心关于发布《化学仿制药参比制剂遴选申请资料要求》的通告(2020年第32号)的要求进行撰写申报资料。
申请资料主要包括以下四个部分:
(一)申请综述:
包括药品通用名、活性成分、剂型规格、适应症简述、药典收载情况、参比制剂发布情况及原研信息等基本信息。
(二)调研信息:
需系统梳理国内外上市情况,包括:
l 国内进口与国产药品批准现状
l 美国橙皮书(Orange Book)收载情况(是否为RLD/RS)
l 欧盟EMA、日本PMDA等监管机构的上市与参比地位信息
(三)调研信息网址链接:
提供所有引用信息的可访问网址,并确保链接有效,便于审核人员核查。
(四)附件(必须包含以下文件):
附件1:化学仿制药参比制剂遴选申请资料自查表
附件2:申报资料真实性声明文件
附件3:拟申请参比制剂可及性证明(如销售数据、采购发票、厂家生产计划等)
附件4:拟申请参比制剂说明书
附件5:相关审评报告或临床研究信息(如FDA审评报告、EMA评估文件等)
附件6:其他支持性资料
填写注意:
1.申请资料中目录及项目编号不得改变,无法提供某一项的相关信息时,该项目的编号和名称也应保留,可在该项下注明“未调研到相关资料”或“不适用”。
2.“四、附件”项下内容可以合并在一个WORD文档内提交,建议大小不超过100M。
3.关键性资料应在对应资料后附中文翻译稿。
四、未通过审议的主要原因分析
CDE公布参比制剂目录未通过审议的主要原因包括:临床安全有效性数据不足、已有更优品种替代、改剂型或改规格无明显临床优势、存在安全性风险、未提供充分的安全有效性数据、与已发布参比制剂重复或浓度一致等。
根据CDE公示数据,近三年参比制剂申请未通过率约35%,主要集中在以下领域:
(一)临床价值不足
证据链缺陷:上市时间较早的品种缺乏现代临床研究数据,无法证实安全性和有效性
剂型无优势:改剂型品种未改变给药途径,生物等效性无显著提升,临床获益不明确
规格不合理:申请规格小于临床最小推荐剂量,或与临床常规用法用量不匹配
(二)安全性风险
工艺缺陷:注射剂采用除菌过滤工艺但可耐受终端灭菌,无菌保障水平不足
辅料问题:含有已知致敏性防腐剂或毒性辅料,无临床使用必要性
质量隐患:生产工艺与注册工艺不一致,存在杂质超标等潜在风险
(三)参比地位不符
非原研申请:申请品种为日本或美国上市的仿制药,不具备参比制剂资格
非主流市场产品:选择未进入ICH成员国市场的品种,缺乏国际公认性
重复申报:申请品种与已发布参比制剂浓度、剂型完全一致,属于重复申请
(四)特殊品种问题
复方制剂:相比单方制剂无明显疗效优势,或未提供协同作用的临床证据
老药再评价:历史数据不充分,需重新开展确证性临床试验
罕见病药物:缺乏足够的临床使用数据,无法评估风险获益比
五、参比制剂目录动态调整机制
调出参比制剂目录的主要原因包括:剂型不合理、不符合现行药典技术要求、已有更优剂型替代、安全性或有效性证据不足等。
根据国家药监局及CDE发布的多批目录调整通告,已发布的参比制剂被调出主要基于以下几类情形:
(一)剂型选择不合理
对于可耐受终端灭菌工艺的注射剂,仍采用冻干粉针等非终端灭菌剂型,被视为不合理剂型。例如,注射用奥扎格雷钠,因其仿制注射液可耐受过度杀灭工艺,原冻干剂型被判定为不合理。
(二)不符合中国药典通则要求
某些吸入制剂未达到“无菌制剂”标准。如吸入用硫酸沙丁胺醇溶液,因不符合《中国药典》关于吸入液体制剂应为无菌制剂的规定而被调出。
(三)缺乏充分的有效性证据
药用炭胶囊因现有资料不足以证明其在急性腹泻及胃肠胀气中的临床有效性,经专家委员会审议后被调出参比制剂目录。
(四)存在更优治疗选择或技术替
当某品种已有质量更稳定、杂质控制更优、生产工艺更先进的剂型上市时,旧剂型可能被调出。例如,终端灭菌注射液在无菌保障和稳定性方面优于冻干粉针,成为首选。
(五)监管政策动态调整的体现
参比制剂目录并非一成不变,而是随科学认知和技术进步动态优化。调出机制旨在引导企业研发更安全、有效、高质量的仿制药,避免重复开发落后剂型。
这一调整机制体现了我国仿制药审评从“有药可用”向“优药优先”的转变,强化了对剂型合理性、生产工艺先进性和临床价值的综合评估。
六、小结:
仿制药的研发发展方向正从“数量扩张”转向“质量优先与临床价值导向”,未通过审议品种增多和参比制剂调出机制常态化,反映出监管层对仿制药立项、研发和申报的高标准、严要求趋势。
参考资料
2023-06-08国家药监局关于发布化学仿制药参比制剂调整程序的公告(2023年第35号)
2022-03-11关于公开征求《已发布化学仿制药参比制剂调整程序》意见的通知
2020-10-19国家药监局药审中心关于发布《化学仿制药参比制剂遴选申请资料要求》的通告(2020年第32号)
2019-03-25关于发布化学仿制药参比制剂遴选与确定程序的公告(2019年第25号)
2016-03-08普通口服固体制剂参比制剂选择和确定指导原则




